
Wykrywanie anomalii to techniki używane do identyfikowania i znajdowania nietypowych wzorców niezgodnych z oczekiwanym zachowaniem. Ogólnie opisałem je w poprzednim artykule. W tym poście przedstawię Wam mój ulubiony algorytm do wykrywania anomalii.
Las izolacji (Isolation forest) – czym jest?
Isolation forest został zaproponowany przez Fei Tony Liu, Kai Ming Ting i Zhi-Hua Zhou w 2008 roku (link do pracy). Jest to nienadzorowany algorytm uczenia się należący do rodziny drzew decyzyjnych.
To podejście wykorzystuje fakt, że anomalie mają wartości zmienne bardzo różniące się od wartości normalnych. Algorytm działa świetnie z bardzo dużymi zbiorami danych wymiarowych i okazał się bardzo skutecznym sposobem wykrywania anomalii.
Las izolacji – idea działania
Algorytm lasu izolacji wykorzystuje podstawowe właściwości anomalii:
- jest ich niewiele i występują dość rzadko (inaczej nie byłyby anomaliami🙂),
- są inne – mają wartości lub atrybuty mocno różniące się od pozostałych wartości.
Te dwie własności pozwalają na wyizolowanie anomalii z pozostałych danych.

Rdzeniem algorytmu jest „izolowanie” anomalii poprzez tworzenie drzew decyzyjnych z losowymi atrybutami. Losowe partycjonowanie daje zauważalne krótsze ścieżki dla anomalii, ponieważ mniej wystąpień (anomalii) skutkuje mniejszymi partycjami. Brzmi skomplikowanie? To prześledźmy krok po kroku prosty przykład.

Las izolacji – opis algorytmu
Isolation forest jest bardzo podobny do Random forests i jest zbudowany w oparciu o zestaw drzew decyzyjnych dla danego zbioru danych. Istnieją jednak pewne różnice. Isolation forest identyfikuje anomalie jako obserwacje o krótkich średnich długościach ścieżek na drzewach izolacyjnych. Dla każdego drzewa izolacyjnego stosowana jest procedura:
- Wybierz losowo dwie funkcje.
- Podziel punkty danych, losowo wybierając wartość między minimum a maksimum wybranych cech.
- Podział obserwacji jest powtarzany rekurencyjnie, aż wszystkie obserwacje zostaną wyizolowane.
Spójrzmy na prosty przykład, gdzie mamy tylko dwie funkcje (wzrost i waga) oraz 6 zmiennych.

Jako pierwszy krok wybrano zmienną „waga” i wylosowano wartość 110. Jeśli obserwacja ma większą wartość niż 110, to zostaje oznaczona jako wartość odstająca (czerwony kolor). Patrząc na powstałe drzewo można wyróżnić normalne punkty i anormalne na podstawie ich średniej długości ścieżki. Im krótsza droga do wyizolowania, tym bardziej wskazuje to na anomalie.
Pamiętajmy, że to tylko jedno drzewo. A jak sama nazwa wskazuje ma to być las, czyli zbiór mnóstwa drzew.
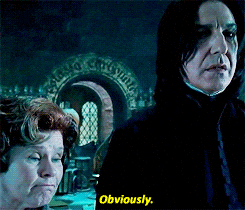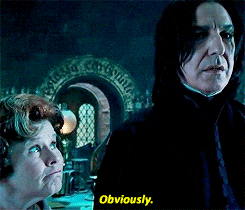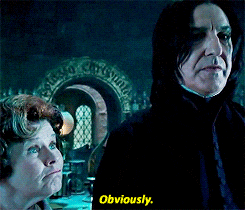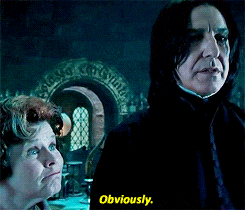
Skoring anomalii (Anomaly score)
No dobrze, ale jak na podstawie tych wszystkich drzew powiedzieć, czy coś jest anomalią czy nie?

Nie tylko las izolacji, ale każdy algorytm wykrywania anomalii musi ocenić swoje punkty danych i określić „zaufanie”, jakie algorytm ma do potencjalnych anomalii. Wygenerowany wynik anomalii powinien być ograniczony i porównywalny. W lesie izolacji fakt, że anomalie zawsze pozostają bliżej korzeni jest naszym głównym puntem zaczepienia, który pomoże nam zbudować funkcję punktacji. Nazwiemy ją scoringiem anomalii.
W uproszczeniu jest to funkcja, która liczy średnią długość ścieżki od wierzchołka. Autorzy algorytmu zaproponowali, aby jego wartości mieściły się w przedziale od 0 do 1 (choć mogą być różne implementacje tego algorytmu, np. jak w scikit-learn anomalia jest dla wartości mniejszych od 0, a większe to wartości normalne).
Wynik anomalii jest zdefiniowany jako:

W powyższym równaniu h(x) reprezentuje długość ścieżki punktu danych x w danym drzewie izolacji. Natomiast wyrażenie E(h(x)) oznacza wartość oczekiwaną lub „średnią” wartość długości ścieżki we wszystkich drzewach izolacji. Wyrażenie c(n) reprezentuje średnią wartość h(x) przy danej wielkości próbki (w tym przypadku n-razy).
Po obliczeniu wyniku anomalii s(x, m) dla danego punktu otrzymujemy wartość pomiędzy 0 a 1. Możliwe są takie sytuacje:
- Gdy wynik obserwacji jest bliski 1, długość ścieżki jest bardzo mała i wtedy punkt danych można łatwo wyizolować – mamy anomalię.
- Gdy wynik tej obserwacji jest mniejszy niż 0,5, długość ścieżki jest duża i wtedy mamy normalny punkt danych.
- Wszystkie obserwacje mają wynik anomalii około 0,5, czyli cała próba nie ma żadnej anomalii.
Przykład w Python
Krok 1 – wczytanie i przygotowanie danych
Wykorzystamy znany Wam z różnych tutoriali zbiór Boston
import numpy as np
import pandas as pd
import seaborn as sns
import plotly.express as px
import matplotlib.pyplot as plt
import shap
from sklearn.datasets import load_boston
from sklearn.ensemble import IsolationForest
boston = load_boston()
df = pd.DataFrame(data=boston.data,columns=boston.feature_names)
df['target']=boston.target
df.sample(5)

Zbiór składa się z 14 zmiennych:
- CRIM – wskaźnik przestępczości na mieszkańca według miasta
- ZN – część działki pod zabudowę mieszkaniową pod działki o powierzchni ponad 25 000 stóp kwadratowych
- INDUS – odsetek niedetalicznych akrów biznesowych na miasto
- CHAS – zmienna Charles River (1, jeśli trasa ogranicza rzekę; 0 w przeciwnym razie)
- NOX – stężenie tlenków azotu (części na 10 milionów)
- RM – średnia liczba pokoi na mieszkanie
- AGE – odsetek jednostek zajmowanych przez właścicieli wybudowanych przed 1940 rokiem
- DIS – ważone odległości do pięciu centrów zatrudnienia w Bostonie
- RAD – indeks dostępności do radialnych autostrad
- TAX- pełna stawka podatku od nieruchomości od 10 000 USD
- PTRATIO – stosunek liczby uczniów do nauczycieli według miasta
- B – 1000 (Bk – 0,63) ^ 2, gdzie Bk to odsetek czarnych według miasta
- LSTAT -% niższy status populacji
- MEDV – mediana wartości domów zajmowanych przez właścicieli w tysiącach dolarów
fig, axs = plt.subplots(ncols=7, nrows=2, figsize=(10, 5))
index = 0
axs = axs.flatten()
for k,v in df.items():
sns.boxplot(y=k, data=df, ax=axs[index])
index = index + 1
plt.tight_layout()

Dzięki wykresom pudełkowym od razu widzimy w ujęciu pojedynczych zmiennych wartości odstające, na przykład:
- dla zmiennej CRIM wartości powyżej 10,
- dla zmiennej CHAS wartość równa 1,
- czy dla zmiennej B wartości poniżej 350.
W zbiorze zmienną celu jest wartość mieszkania. Wyciągnijmy tylko zmienną, aby nie zaburzała obrazu i na pozostałych zmiennych sprawdźmy, które obserwacje zostaną zakwalifikowane jako anomalie wg algorytmu isolation forest.
X = df.drop('target', axis=1)
y = df['target']
Krok 2 – przeliczenie isolation forest
Zobaczmy najpierw jakie mamy najważniejsze parametry:
- n_estimators – liczba drzew wykorzystanych do lasu izolacji,
- contamination – parametr mówiący o proporcji wartości odstających w zbiorze. Może przyjąć wartość „auto” lub liczbę rzeczywistą pomiędzy 0 a 1. W przypadku ustawienia jako „auto” poziom zostaje ustawiony na podstawie zaimplementowanego wzoru z pierwotnej pracy naukowej,
- max_samples – liczba wierszy brana do pojedynczego drzewa z wszystkich wierszy. W przypadku „auto” warto mieć świadomość, że dla większych zbiorów zostaną wzięte tylko 256 wiersze (max_samples=min(256, n_samples)),
- max_features – maksymalna liczba zmiennych cech do szkolenia estymatora.
Zobacz na poniższym przykładzie, jak zmiana parametru contamination wpływa na zmianę outlierów:
for contamination in ['auto', 0.10, 0.05, 0.03, 0.01]:
iforest = IsolationForest(n_estimators=500,
contamination=contamination)
iforest.fit(X)
y_pred = iforest.predict(X)
df['scores']=iforest.decision_function(X)
df['anomaly_label']=y_pred
print(f'For contamination={str(contamination)} \
predicted outliers: {df[df["anomaly_label"]==-1].shape[0]}')

Ja chciałbym, aby moje anomalie stanowiły maksymalnie powiedzmy 5%. Zatem wytrenujmy nasz ostateczny model i prześledźmy wyniki.
iforest = IsolationForest(n_estimators=500, contamination=0.05)
iforest.fit(X)
y_pred = iforest.predict(X)
df['scores']=iforest.decision_function(X)
df['anomaly_label']=y_pred
print(f'Outliers: {df[df["anomaly_label"]==-1].shape[0]}')
df[df.anomaly_label==-1].head()

Od razu rzucają mi się w oczy wartości, które były wartościami odstającymi w naszych wykresach pudełkowych. Tylko, że w tym przypadku często kilka z nich występuje dla pojedynczego wiersza.
Krok 3 – Interpretacja i zrozumienie modelu
W pierwszym kroku przyjrzyjmy się rozkładowi wyliczonemu spoza anomalii:
df['anomaly']=df['anomaly_label'].apply(lambda x:
'outlier' if x==-1 else 'inlier')
fig=px.histogram(df,x='scores',color='anomaly')
fig.show()

W sklearn mamy implementacje isolation forest, gdzie wartości są traktowane jako odstające dla scora poniżej 0. Gdybyście chcieli zwiększyć lub zmniejszyć liczbę wartości zaklasyfikowanych jako anomalie, to zamiast zmian na parametrze contamination, możecie dobrać własny próg odcięcia bazując na scorze.
Ciekawym pomysłem jest zwizualizowanie pojedynczego drzewa.
from sklearn.tree import export_graphviz
from subprocess import call
from IPython.display import Image
estimator = iforest.estimators_[1]
#print(estimator)
export_graphviz(estimator,out_file='tree.dot',max_depth=5,
feature_names = X.columns,filled = True,
special_characters=True,rounded=True,precision=2)
call(['dot', '-Tpng', 'tree.dot', '-o', 'tree.png', '-Gdpi=600'])
Image(filename = 'tree.png')

W powyższym wykresie widzimy najkrótszą ścieżkę dla cechy RM pomiędzy 4.23 a 4.38, która łapie tylko jedną obserwacje ze zbioru.
Niestety mamy w naszym modelu 500 takich drzew. Dalsze ich wizualizacje nie doprowadzą nas do lepszego zrozumienia jak działa model, więc lepiej pójść w innym kierunku.
Wykresy rozrzutu
Aby lepiej rozumieć model, możemy się przyjrzeć jeszcze wykresom rozrzutu z dwiema cechami jednocześnie.
fig = px.scatter(df,x='AGE',
y='CRIM',
color='anomaly')
fig.show()

Tutaj można zauważyć, że większość anomalii dotyczy starszych budynków. Dodatkowo, jeśli budynek jest stary i wskaźnik przestępczości na mieszkańca wynosi powyżej 40, to zauważ, że w prawej górnej ćwiartce mamy prawie same anomalie.
Analizując takie wykresy nabierzemy intuicji, na co spojrzał nasz model.
T-sne
Możemy jeszcze spojrzeć na więcej cech na raz korzystając z PCA (szczegóły) lub t-SNE (szczegóły), aby sprawdzić, czy wg tych metod punkty anomalii są blisko siebie.
from sklearn.manifold import TSNE
X_tsne = TSNE(learning_rate=100, perplexity=50).fit_transform(X)
df['tsne 0'] = X_tsne[:,0]
df['tsne 1'] = X_tsne[:,1]
fig = px.scatter(df,x='tsne 0',
y='tsne 1',
color='anomaly')
fig.show()

Nie ma jednoznacznego fragmentu na wykresie, który skupia anomalie. W tym przypadku interpretuję to w ten sposób, że model lasu izolacji spojrzał wielowymiarowo i wyszukał różne anomalie ze zbioru danych nie koncentrując się tylko na kilku zmiennych. Ta wizualizacja upewnia mnie, że model fajnie spojrzał całościowo na dane.
No dobra, tylko jak jeszcze lepiej zrozumieć model? Jak nie wiesz jak, to wykorzystaj mój ulubiony pakiet, czyli…
Shap
W tym artykule opisałem dokładniej jak wylicza się wartości Shapleya i je interpretuje. Dlatego nie będę się rozdrabniał i z grubsza opiszę wykresy.
Przeliczmy shapa…
explainer = shap.Explainer(iforest, X)
shap_values = explainer(X)
shap.initjs()
I zacznijmy od wyjaśnienia konkretnej obserwacji (np. 490):
i=142
shap.plots.force(shap_values[i])

Zmienne pokolorowane na czerwono pokazują, w jaki sposób przyczyniają się do zwiększenia wartości predykcji. Kolor niebieski wskazuje, w jaki sposób cechy przyczyniają się do zmniejszenia prognozy. W naszym przypadku cechy zaznaczone na niebiesko są wyznacznikami znalezienia anomalii. Długość każdej strzałki przedstawia wpływ tej funkcji na prognozę.
Widzimy, że w przypadku obserwacji (wiersza) numer 490 na bycie anomalią wskazują przede wszystkim wartości dla zmiennych INDUS, TAX oraz LSTAT.
Możemy na tą samą obserwacje spojrzeć jeszcze na wykresie wodospadowym „waterfall”, który moim zdaniem jest prostszy i czytelniejszy.
shap.plots.waterfall(shap_values[i])

Całościowe spojrzenie na cechy pokazuje nam, które charakterystyki całościowo najmocniej wpływają na oznaczenie obserwacji jako anomalii i dla jakich wartości:
shap.summary_plot(shap_values, X)

Dla przypomnienia: cechy na tym wykresie są uporządkowane na podstawie sumy bezwzględnych wartości Shapleya. Zatem im wyżej na wykresie, tym cecha bardziej przyczynia się do wyniku modelu. Natomiast różne kolory wskazują wartości funkcji w zestawie danych.
Najmocniejszymi cechami są:
- ZN – niższe wartości (kolor niebieski) wpływają w ten sposób, że większe jest prawdopodobieństwo określenia obserwacji jako anomalii (bardziej na prawo na wykresie),
- CRIM – niższe wartości to większa szansa na anomalie,
- B – wyższe wartości wskazują anomalie.
Gdybyście chceli kopać głębiej zawsze można zobaczyć jak wygląda pojedyncza cecha np. ZN czy AGE:
shap.force_plot(explainer.expected_value, shap_values, X)

Głównie za anomalie odpowiadają wartości ZN do 10.

A dla wieku zwiększa się prawdopodobieństwo sklasyfikowania jako anomalii od wieku około 60 lat.
Podsumowanie
Mam nadzieję, że od teraz wykrywanie anomalii nie będzie Wam straszne. I dodatkowo, że będziecie umieli zinterpretować, dlaczego Wasz model podjął taką, a nie inną decyzję.
Pozdrawiam serdecznie z całego serducha,




Mam wrażenie, że w interpretację summary_plot wkradł się błąd. W sklearnowym isolation forest ujemne wartości to anomalie, czyli to, co będzie na lewo na wykresie (ciągnące w dół) będzie zwiększało prawdopodobieństwo anomalii, a to, co na prawo będzie zwiększać szansę na wynik „typowy”.
Czyli „ZN – niższe wartości (kolor niebieski) wpływają w ten sposób, że większe jest prawdopodobieństwo określenia obserwacji jako anomalii (bardziej na prawo na wykresie)” –> niższe wartości (niebieski) to niższe prawdopodobieństwo anomalii, bo „ciągną wynik w górę” czyli do wartości częstych.
co rozumiesz przez określenie inliner?
Zawsze spotykałem się z tym, że to określenie tyczy się błedów w danych ukrytych w wśród normalnego rozkładu danych.
Hej Dawid!
Jeśli masz na myśli wykres, gdzie oznaczyłem cos albo jako „outlier” albo „inlier”, to po prostu użyłem słowa jako wyraz odwrotny do outlier – nic więcej. Nie słyszałem takiego znaczenia tego słowa. Dziękuje za informacje 🙂
Gdybym nie miał tyle wykresów do poprawienia to zmieniłbym teraz na „normal” by nie wzbudzać niepotrzebnych pytań 😉
Podstawiam serdecznie,
Mirek